


A Defense for the Accuracy of the Specificity of Sars-Cov 2 Sequencing
Part 1?

THIS BEGAN AS A DM THREAD, SO SORRY FOR TYPOS, AND THE ODD NATURE IN WHICH IT BOUNCES AROUND. IT SHOULD STILL BE VERY USEFUL FOR PEOPLE WHO ACTUALLY WANT TO UNDERSTAND THESE TECHNOLOGIES. THE LINKS ARE NOT FULLY SOURCED IN THE FOOTNOTES AS A RESULT HOWEVER, SO RECORD THEM IF YOU NEED THEM PERSONALLY, IN CASE THE LINKS ARE EVER BROKEN.
Sequencing is decentralized. Many scientists perform it all around the globe. It's really the most decentralized data point we have when it comes to finding nucleic acids in the natural world. That's many thousands of eyes looking it over for errors. I can't talk about types I do not understand but I can talk about a couple that I keep bringing up. Most importantly, Sanger.
Sanger works like this:
First off, nucleic acid (usually from biological samples) chemical amplification of source material via PCR is done, (see ”final thought experiments” section, to learn about RNA to cDNA reverse transcriptase reactions and RT-PCR) to generate enough material off the sample to then sequence. That alone is enough to probably make some individuals hate it, but again, properly created primers bind to genetic material via the complementary-base-pairing-rule, and then amplify a target region via a Polymerase "chain reaction". This is necessary because each base in the source material genome will be determined many times over (coverage?) with a separate molecule.
The primer is like a catch point for the viral ribonucleic acids onto which the polymerase will then attach and amplify the molecules after the primer of the original material (only needing to be 20 nucleotides long usually, with multiple different primers used to amplify many regions after them across the genome, so again, this should put into perspective the statistical power of identification behind a nucleotide sequence of that length. And no, the amplification doesn't invalidate anything, as long as you measure the human material versus the material that is then generated, as PCR is a measurable process of amplification that doubles the material being amplified each time, until you run out of reactants {2x2x2x2 but with latched on DNA or RNA molecules}. Matter can't be created or destroyed. The measurability of this process is also how differences in source nucleic acids can be inferred with the tests i.e. if it takes me 40 cycles to generate the same amount of viral material from a good set of conserved influenza primers, yet only 20 to get the same amount of material for SARS CoV 2, from 1/2 each of a patient sample, then I know I have many times the amount of SARS 2 RNA, than I do influenza RNA in the sample. Quantify the human DNA/RNA too and you can know the exact ratios of everything present in the sample. Viral material isolation is also an important concept to know
I'd like to reiterate now that PCR just stands for Polymerase “chain reaction”, if it's not clear how exact the math behind one is when it's working right. Does this have a slight error rate? Yes, but, it should be completely measurable. If those (polymerases) didn't work, life wouldn't work. This is why it's generally so accurate if the primer is correct.
After this, sanger is then performed on the amplified sample cDNA (probably after splitting the strands into single strands with NaOH [Sanger full reaction breakdown] Denaturation of nucleic acids), which ALSO uses a PCR (again, a measurable chemical reaction [Balancing chemical equations]) but this time a special one, called chain termination PCR.
In chain-termination PCR, the user mixes a low ratio of chain-terminating ddNTPs in with the normal dNTPs in the PCR reaction. ddNTPs lack the 3'-OH group required for phosphodiester bond formation; therefore, when DNA polymerase incorporates a ddNTP at random, extension ceases. The result of chain-termination PCR is millions to billions of oligonucleotide copies of the DNA sequence of interest, terminated at random lengths (n) by 5’-ddNTPs.
In manual Sanger sequencing, four PCR reactions are set up, each with only a single type of ddNTP (ddATP, ddTTP, ddGTP, and ddCTP) mixed in. In automated Sanger sequencing, all ddNTPs are mixed in a single reaction, and each of the four dNTPs has a unique fluorescent label.
Sanger Sequencing Steps & Method
This causes random fragment lengths to be created, which are likely tweakable by the amount of special terminating molecules you added. I don't know the average fragment length, but if it's just 6 or more, we have now reached two Amino_acids, and let's not forget the primer itself (from the origin sequence) is usually 20nt long (6 amino acids in a coded protein, if all were used for that function). So I'm guessing the average length of the fragments is pretty big (21-idk, 30 or more?).
This video of Sanger sequencing shows it visually well. Next the bases in a Sanger sequence are determined by the speed at which they pass through the Electrophoresis gel, and where they settle. This is determined by the atomic weights of the atoms in the base, and is backed up by verifiable chemical maths (here is a random example of molar mass and moles). That is the basis of all good science (atomic/molecular weight measurements of elements/compounds). They pass through the gel at a rate that is inversely proportional to their molecular weight (sum of the atomic weights of all the atoms in the base) which enables the fragments to be separated at a resolution of 1 base. The strand passes through a laser, which hits the special terminated base that usually has a dye in it, emitting a corresponding wavelength of light to identify what the base at the end of the strand is, which they call a "base call". This gives you a pool of fragments, and you know exactly what the bases are at the end of each strand. These can then be mapped against the reference genome, which is where that picture I posted below comes in, and you have now mathematically verified the presence of all the viral RNA molecules you were looking for.
Added note: the primer is determined from the reference genome. This tech is giving you information about what the exact end base is of each fragment, and the separation is ordering the fragments by length perfectly (1 base resolution). But it's the primer, then difference of extension, that is creating this perfectly recorded order of what is there, 1 nucleotide at a time. It's very hard to visualize, but let's say those first 30 were the primer (that was based literally from the already known reference. that's why in an interview, Kevin Mckernan once said "you always need to know a little bit of sequence, to get a little bit). Let's say we then have a polymerase latch on, then start adding complementary bases, until it comes to a random stop. Now let's imagine there are millions of that first primer, grabbing everything available that starts there, i.e. millions of different molecules with that starting primer (and remember, we amplified the same sets of molecules from PCR, thanks to their primer binding), and that we have termination bases with dyes for all 4 nucleotides. Then let’s imagine, we have polymerases grabbing and stopping the reaction, at each and every nucleotide on different strands. So, starting from those first 30, we are getting fragments that stop at each base after it, and they end up separated by length of 1 base, and we know the end base of each fragment. It's walking down the genome that's present and verifying what is there, using a fragment to determine each end base. Despite this happening with millions of fragments, the nucleotides are still so exact off the primer, that they end up nearly perfectly matching the reference consistently. However we actually use many primers all down the reference genome too (and get additional coverage/verification of the same sections, with other strands). I'm sure sanger lengths don't go too long before they terminate, so this is why you will need a lot of primers from your known reference. This is why sanger can be used to "verify the bases' ' a PCR test amplifies, it pretty much checks the entire genome of what was amplified, especially what was in between the original PCR primers. The sanger primers bind, and then we get to know every nucleotide after, with a copy of each amplified molecule being used to tell us each base.
This is the only point I find confusing, as I’ve never done Sanger or these lab practices hands on:
How much coverage is done? How many molecules are assessed, and do you determine a consensus average from those? I would think this is subjective and vulnerable to tuning, by creating more material from your first RT-PCR, or perhaps by performing standard PCR after the RT-PCR, to generate even more cDNA. It’s all really POINTLESS though, as people are arguing against the viability of these techs to determine the presence of viral particles, and if those particles were a likely source of infection. If PCR primers are accurate (something anyone with the desire to look at genomes and corresponding virus primers can determine) then they grab the right viral molecules. If they grab the right molecules, the polymerase amplifies everything after that consistently. Sanger then verifies this back to the reference using all amplified particles, (that were each only chosen by the initial primers, or they would not have amplified at all) consistently. These genomes can be recorded, then transfected into new cells after conversion back into RNA from cDNA, then nucleic acid codes produced from those cell culture transfections, and verified back to the reference. Which is also done, consistently:
infectious cDNA clone of SARS CoV 2
For the: “surely these infectious clone particles are more pure than the natural virus”? crowd. First, the above paper shows that they are not. Second, the “natural virus” is maintained despite its mutative quasiswarm state, by a concept known as Negative selection. When negative selection is behaving appropriately, it pushes quasiswarm clouds towards Stabilising selection. This is why the mutation rate of any endemic virus already known to maintain its own spread (remember, sequencing is NOT bunk and IS decentralized) is NOT SYNONYMOUS with the rate of virus mutations that will stick in the swarm. Viral particles that can’t replicate due to mutation, remove themselves from the swarm due to this inefficiency, ensuring only that which is viable can reproduce, and thus go on to infect another host.
Lets also talk about the different ratios of proteins (having more S protein sub-genomic mRNAs than N, and much more of those RNAs than full viral genomes, a critique sometimes lobbed at nanopores that don’t find enough full reads to satisfy certain individuals):
This is just a miscomprehension of what the virus actually does to construct itself, and this is where the only gray comes in that I can see. It's also irrelevant gray, because the physical quaternary code that is Sars 2 can be consistently found and documented with existing tech, proving its ability to maintain itself, in everything from people to cell cultures. But, the more I think about this, the more I realize Kevin's statement is probably the best explanation of it. If I have 1 viral genome, yet I need to package that genome in N proteins, and then envelope proteins, then I need to put multiple spike trimers with 2 subunits all around this particle, then it stands to reason I would need more sub-genomic RNAs to make the right distributions of proteins to nucleic acids. There are also likely poorly understood alternate functions of some of these proteins within the cell, and as for full genome reads, those are nanopore snapshots in time, those are probably continually ejecting those full reads at a somewhat steady rate during proper infection. A virus needs a good balance of replication to spread, it's not just going to copy the crap out of itself and destroy the host, if it does its an evolutionary dead end, and it as such it stands to reason full viral particles would be rather short lived, ejected quickly and out to do their job quickly, and not at too fast a rate, as the immune system is actively hunting those particles through Chemotaxis. Too many virions too fast, and you hurt your host as well as prompt the immune system faster. At least this is what I think about the gray area, but I still again, see it as irrelevant to the discussion of whether or not RNA viruses can maintain themselves, since primers and polymerases work on complementary base pairing, and stuff like sanger/nanopores are so accurate
BACK TO SEQUENCING AND THE “CHUNK REARRANGEMENT, PUZZLE PIECE” ARGUMENTS
So, what's the point in all that? Well, some people like to argue that perhaps these rearranging chunks are being "rearranged into something else''. This is the part I can't seem to get people to understand. An amino acid, is coded for by a codon triplet from genetic code, when it passes through a ribosome. There are also start codons/stop codons, untranslated regions that do other stuff etc. but that's irrelevant for what I'm trying to portray. So let's say, Sanger grabs a, idk 30bp chunk of genetic material. I'll pull one from Sars CoV 2 now. “attaaaggtt tataccttcc caggtaacaa”
In sanger, these would be verifiable based upon the atomic weights that separated the fragments and ordered the colored terminated bases. Those are the first bases in the SARS CoV 2 consensus sequence (which probably makes them a bad example. I have a feeling they may need to be a primer, unless you use a primer ahead of this spot, which makes a polymerase go backwards. Might be possible, expert needed for this question.) Now, I'll bet you 100$ that you can take that chunk, and start looking for it in influenza, rhinovirus, human genomes etc, and that you would almost never find it in anything else. The reason behind this is because of the probability that these identical bases will show up again. They are all dependent on the prior acid being right, which is a form of dependent probability.
So, how do we calculate probability like this? It would be (1/4 x 1/4) 30 times. That reaches percent chances that they show up by chance somewhere else, into numbers so very small they are difficult to even comprehend. Now, it's not quite this simple, as recombination can occur (making this chunk move to another virus perhaps, or perhaps it's from a gene that all viruses have to use a lot. That is what I mean when I say conserved sequence because some genes CAN'T change a lot, as evolution or life barriers prevent this. A good example is the RdRp gene in coronaviruses, so maybe large chunks of those genes would be in multiple viruses). But, let's remember, that in Sanger, we're breaking up everything, and amplifying it appropriately, probably with a RT-PCR that covers the whole already known genome, like the ARTIC PCRs, or regions that are already known to be conserved to that specific virus (the HIV inserts of the spike protein are actually an AMAZING candidate for this, too bad they are ignored). So we can easily see that the conserved regions would be amplified as long as we used the correct types and amounts of primers, as well as those that frequently mutate and are NOT conserved across viral species, which is the MAJORITY of the genome. So, we can easily, upon amplification, see every single part of the genome, and we can then map it back to the reference, showing all the chunks are easily present. The accuracy of those genomic codes is such that you can't take these large chunks, and confuse them for other viruses if you are assessing all (or even most) of the material in the sample. It's like, trying to take a single sentence from anywhere in this giant explanation, and looking for it in other literature. You're not going to find it. Maybe you'll find some of it? But if you broke all my sentences up into half sentences, you still could never rearrange it into another piece of literature. In order to do that, the chunks would have to be way smaller, only 3bp (gene specific primers are usually 20bp minimum), so you could rearrange the amino acids into any order you saw fit.
Another important point is that all over the world, scientists are doing these experiments, many times with different primers that are still from various parts of the reference, yet still managing to consistently amplify the same material.
Forgot this. This is for visualization of the genome chunks against the reference. This is not the same kind as Sanger though, which I need to make clear. I'm pretty sure for Sanger, you'd only have 1 primer sets throughout the reference (not sure though), rather than these double primers, as you separate by length and then record each nucleotide on one end of each fragment in Sanger. It's still good for helping to visualize, if you just ignore the fact that there is a second primer on a lot of these. This is probably for something I haven't learned well yet, but it also is still probably very good proof of the accuracy of the reference since as you can see, there are primers across pretty much the whole reference, and we have amplified center regions that also do cover nearly the whole reference (except maybe the very beginning of the reference, which is why I said the first thirty were a bad example, as they may have to be a primer, since in the below picture this is all started at the 5' end of the virus to then start recording what is after the beginning of the fragment. This is obvious because there is no blue coverage for the very first part of the virus in the picture below, only red primer coverage
[ Directionality ]
I think this is something like illumina, not Sanger. However it may be possible to get some types of polymerase to go the other way, up the genome as well, which is how I’m assuming the poly A tail primers or even random hexamer primers may work, so that critique may not even be accurate, I’m simply basing it on the picture below. however I do know DNA polymerases are limited by directionality, so this is a biochemical gray area for me. I’m confident though, that following it to its origin and factual nuances will only unearth more viable, well tested science). After Sanger or another sequencing tech is used to identify amplified regions of new viruses, primers that span novel regions of a novel viral genome can be created, enabling back testing of said discovered genome, and then those amplified products can also be Sangered, verifying the validity of your novel, gene specific primers.
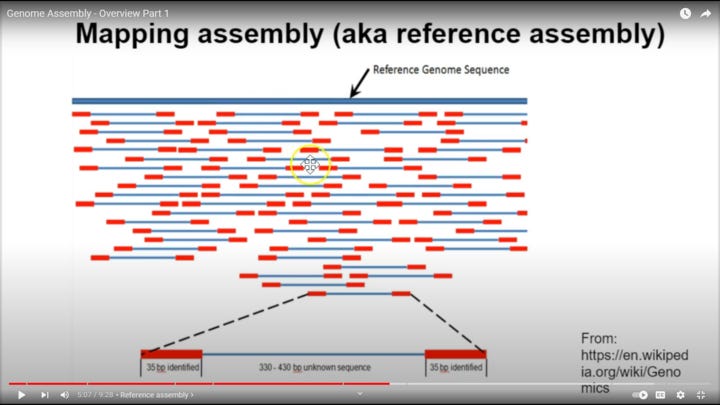
The most important point we need to remember of all this though, is we're asking about "how accurate was the reference genome? Obviously very accurate, or the primers would not even succeed in grabbing the material in the first place. In the above picture, we can see there is actually primer coverage for the entire genome across all the chunks (red sections).
As I understand them, these techs take human samples, then with primers that are based from the reference genome (in pretty big chunks, usually 20 bases in a row or more, which is enough to code for 6+ amino acids), bind and amplify what is there ONLY IF if the primers succeeded in chemical binding of DNA/RNA strands present in the sample (if from an RNA virus, also converting to cDNA, or if DNA virus, perhaps splitting the center bond to allow primer binding). This binding and subsequent amplification will only happen if complementary strands are present to initiate the reaction, or if human/mechanistic error occurs. Then in Sanger, they are then extended off those primers, and terminated at 1 base lengths with special dyed bases that stop the polymerase reaction, enabling recording of the whole genome that is actually amplified from the material. In nanopore, as was shown by Kevin, a sample can be assessed with none of these needed steps. Sanger is great for saying "is this chemically verifiably there" and "is it different than my current record of it". That, correlating with sick people out of a hospital, or from their snot you put into a plate (most likely after viral isolation), which started killing cells on that plate that otherwise are fine, is incredibly strong proof of infection, especially if you take other primers from other viruses, and also check said samples for them (as well as use random hexamer primers and oligo(DT) primers, which anneal to poly A tails).
Nanopores show us the entire quasiswarm, and yes, there are other viruses present, but we can also assess what percentage at which they were present in the samples
From kevin's article:
Quasi-Species Swarms in SARs-CoV-2 - by Anandamide
Most notable from this work is that by sequencing all RNA in patient samples, we can shed light on the blame game. Many people like to claim C19 is just a passenger and something else is causing illness. But that something else has no RNA signatures in Butler et al. Only 3.2% of the samples had co-infection with other RNA based viruses and in many patients the C19 RNA was the dominant RNA in the sample, even more prevalent than host cell RNA
It's very hard to put into perspective the breadth of knowledge that lies underneath microbiology, from the history of basic chemistry and the discovery of elements Introduction to chemistry | Atoms, compounds, and ions | Chemistry | Khan Academy RutherfordÂs gold foil experiment | Electronic structure of atoms | Chemistry | Khan Academy
all the way up to the application of those principles in the field, that enable us to say the things we do about viral theories
The discovery of the double helix structure of DNA Hershey and Chase conclusively show DNA genetic material
Stuff like measurable chemical reactions based upon the properties contained with elements from the periodic table, even down to the machines used to infer results, being based off of molecular physics principles, like inverse proportionality in electrophoresis. It's less that it's on such "shaky ground" (which some end aspects well could be) but more that the building blocks of knowledge under it are so vast, it's very easy to see grey where we don't understand all the steps.
Final thought experiments and information concerning the RNA to DNA step:
Before the sequencing Sanger step, if dealing with an RNA virus, I’m 99% sure you would have to convert your RNA material into cDNA first. So rt-PCRs and various known or conserved primers could be used to generate ratios of material off a sample, and then the codes converted to cDNA, to then have the cDNA broken apart by applying NaOH to it, splitting it into two strands, so a DNA polymerase can then come in and apply the special dyed reaction stopping bases, telling you the code of the originally amplified source material. I have a feeling, this is may also be a reason why genome codes for RNA viruses are usually in ATCG, despite RNA actually being AUCG https://en.wikipedia.org/wiki/Uracil
However I don't know this for a fact (Double stranded RNA does exist, though I doubt it is used much, if at all. Specifics of chemistry for RT PCRs can be found here Chemistry of RT-PCR) I've just never seen a Sanger paper use RNA to my knowledge, and I've never seen a RNA genome recorded with U's, it's always T's, indicating the information is nearly always coming from DNA when we actually do the sequencing, and this is the process I know of that reverse transcribes RNA. But again, complementary pairings is how these polymerases/primers work. They are well studied and the different types used can be found/researched using the links in this explanation. I would not be surprised to find out these error rates are so well studied, we know them for each polymerase frequently used. And as usual, as long as we are accounting for the mass of all the molecules originally there, and those that were added, then we know our original code primers, inferences into the produced sequence order, and even inferences into the ratio of sample that code held (and other codes held, like trying to amplify influenza primers for example, as a control), are correct. Best way to do an investigation knowing all this, in my mind, would be to split a sample in half. Do your investigatory steps with poking various rtPCRs to amplify known conserved regions of many viruses, on one half of the sample. Then, take the most suspect one that was amplified (lower CT count, especially if it is more than host RNA/DNA) and figure out what their bases were after the primers, with Sanger. Once you establish a sequence with sanger, make PCRs to amplify other novel regions you discovered via Sanger. Quantify host RNA/DNA against your new sequence primers in the other half of your sample, using your newfound primers to amplify via rtPCR this time, then Sanger again, and verify it was dominant in the other half of the sample too. At least that's what I would do, if these were my only tools, and I'm pretty confident, if I spent enough time making sure my mass was accounted for and all my amplification CTs/polymerase error rates were right, that I could possibly pin down a new virus in a sample in this manner.
This is all possible thanks to this simple indisputable fact: Conservation of matter
Imagine stuff like that, but with many people all poking samples from people all over the world, with different primers targeting different parts of the reference, and yet, all amplifying the exact same code over and over again from samples from sick people. Add nanopores, and I see no reason denovo is used for anything, other than the fact that it's probably pretty accurate, and easier to do than all this pain in the butt stuff I covered, or this other pain in the butt stuff would have sussed out its incompetence as investigatory tech very quickly. I'm pretty sure, the CDC sequences something like 10% of all samples, just to start with. Add all the other labs in the entire world, doing these experiments, and to me it becomes the best data point I see in the pandemic, the only thing I actually know to be true: i.e. that the material is there, consistently
This RNA to DNA step is likely covered well here, will learn more about it soon: RNA to cDNA via reverse transcriptase
This is likely where rt- PCR would have to be deployed, as the rt in that tech stands for "reverse transcriptase", which is the enzyme/polymerase responsible for this particular process. As usual, it likely has a well studied and well documented error rate
Important notes on rt-PCR steps:
In RT-PCR, the RNA template is first converted into a complementary DNA (cDNA) using a reverse transcriptase (RT). The cDNA is then used as a template for exponential amplification using PCR. The use of RT-PCR for the detection of RNA transcript has revolutionized the study of gene expression in the following important ways:
Made it theoretically possible to detect the transcripts of practically any gene[16]
Enabled sample amplification and eliminated the need for abundant starting material required when using northern blot analysis[17][18]
Provided tolerance for RNA degradation as long as the RNA spanning the primer is intact[17]
Further notes on these specific Primers :
To initiate reverse transcription, reverse transcriptases require a short DNA oligonucleotide called a primer to bind to its complementary sequences on the RNA template and serve as a starting point for synthesis of a new strand. Depending on the RNA template and the downstream applications, primers of three basic types are available: oligo(dT) primers, random primers, and gene-specific primers
Oligo(dT) primers consist of a stretch of 12–18 deoxythymidines that anneal to poly(A) tails of eukaryotic mRNAs, which make up only 1–5% of total RNA. These primers are the optimal choice for constructing cDNA libraries from eukaryotic mRNAs, full- length cDNA cloning, and 3′ rapid amplification of cDNA ends (3′ RACE). Because of their specificity for poly(A) tails, oligo(dT) primers are not suitable for degraded RNA, such as from formalin-fixed, paraffin-embedded (FFPE) samples, nor for RNAs that lack poly(A) tails, such as prokaryotic RNAs and microRNAs. Since cDNA synthesis starts at the 3′ poly(A) tail, oligo(dT) primers potentially can cause 3′ end bias. RNA with significant secondary structure may also disrupt full-length cDNA synthesis, resulting in under representation of the 5′ ends.
Oligo(dT) primers may be modified to improve efficiency of reverse transcription. For instance, the length of oligo(dT) primers may be extended to 20 nucleotides or longer to enable their annealing in reverse transcription reactions at higher temperatures. In some cases, oligo(dT) primers may include degenerate bases like dN (dA, dT, dG, or dC) and dV (either dG, dA, or dC) at the 3′ end. This modification prevents poly(A) slippage and locks the priming site immediately upstream of the poly(A) tail. These primers are referred to as anchored oligo(dT).
Random primers are oligonucleotides with random base sequences. They are often six nucleotides long and are usually referred to as random hexamers, N6, or dN6. Due to their random binding (i.e., no template specificity), random primers can potentially anneal to any RNA species in the sample. Therefore, these primers may be considered for reverse transcription of RNAs without poly(A) tails (e.g., rRNA, tRNA, non-coding RNAs, small RNAs, prokaryotic mRNA), degraded RNA (e.g., from FFPE tissue), and RNA with known secondary structures (e.g., viral genomes).
While random primers help improve cDNA synthesis for detection, they are not suitable for full-length reverse transcription of long RNA. Increasing the concentration of random hexamers in reverse transcription reactions improves cDNA yield but results in shorter cDNA fragments due to increased binding at multiple sites on the same template (Figure 4).
Moreover, use of random primers only may not be ideal for some RT-PCR applications. For instance, overestimation of mRNA copy number is one concern [5]. A mixture of oligo(dT) and random primers is often used in two-step RT-PCR to achieve the benefits of each primer type. For microRNA (miRNA) expression assays, random hexamers are not suitable and special primers must be designed for reverse transcription of miRNA [6,7].
Gene-specific primers offer the most specific priming in reverse transcription. These primers are designed based on known sequences of the target RNA. Since the primers bind to specific RNA sequences, a new set of gene-specific primers is needed for each target RNA. As a result, more RNA is required for analysis of multiple target RNAs. Gene-specific primers are commonly used in one-step RT-PCR applications. (White paper:Improved one-step RT-PCR system)
Here is an example of mass screening of sanger samples, verifying the accuracy of "SARS CoV 2". There are mutations/ low error rates, but this viral material travels the world, makes people sick, and we know that thanks to this process. The first is Sanger verification of spike only samples (a very adequate chunk of code)
The second should be full genome verification with Sanger
Sanger verification of SARS CoV 2 samples
Full length genomic sanger sequencing and phylogenetic analysis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Nigeria
But let's say, that's not enough for you still. That's the point of a nanopore. A nanopore [Nanopore sequencing] can take all the material in a sample, and record the molecules as they are. It's a newer sequencing tech that can be used to assess every molecule in a sample. These technologies find the molecules as they are, and what do they find? Well in this paper that is consistently unfairly hated on for not having enough "full reads", they find chunks of 1000nt as the smaller ones, and can even find the full reads, i.e. complete virions. The Architecture of SARS-CoV-2 Transcriptome - PMC (I think that's it, been a minute since I looked for it). This should be the one I’m talking about (again I say I THINK, haven’t double checked extensively. The nanopore study I’m alluding to indeed exists if this isn’t it
Granted there are only two full virions in the sample, but that's beside the point (that's a nuanced issue with how viruses build full virions, and what is happening in a cell to achieve that) these 1000nt chunks, and full virions, can be compared to the original reference sequence. And what do they find? Near exact matches to the on record sequence. These methods and codes are so accurate, that we can even use them to quantify the mutations in the quasiswarm, by just visually comparing what we find against the known reference or other recorded variants. They CAN'T be confused or rearranged into other viruses. What can happen, and only with jigsaw techs, is that: say we have a bunch of incomplete viral chunks in the sample, from different virions. They might be rearranged into a VARIANT that doesn't exist as a whole particle, but that's about it. This article might help if I still am not making sense
Quasi-Species Swarms in SARs-CoV-2 - by Anandamide.
This paragraph in particular ( which covers the above paper)
Kim et al. used Oxford Nanopores to sequence the RNA directly without any conversion into cDNA. This platform sequenced the whole virus with 30Kb reads. No fragmentation and assembly were required. You can see a minimum of ~2,000X coverage across the whole virus in Figure 2B and 400,000X coverage on the 3’ end of the virus (right side of the virus). So lots of subgenomic RNA is made and a minority of the transcripts are full length. The paper further itemizes that middle region of 2,000x coverage and finds 111 reads that span the entire viral genome.
So, how on earth, are we going to confuse viruses for something else? Remember that 30nt chunk I posted from SARS 2? I challenge you to take it, and start hunting for it in any other virus. I'd bet you'd never find it, unless it was from a conserved protein or gene, and even then, it won't show up in viral families that are far removed. It's probably not going to show up in bacteria or human DNA. This is the point of "BLASTing" something too. When we BLAST something, since these codes are so accurate, we can see where else, if anywhere, they have occurred in nature. For example, someone in PANDA just blasted our first RnRp primer from drosten’s paper. We can see, out of ALL recorded human genomes in the entire world, it only has a 95% match to a single part of human Chromosome 3, outside of where it is in SARS Cov 2 (and Drosten didn't make the primer match that portion from SARS CoV 2 perfectly either, he is not honest and did something weird). It doesn't show up anywhere else in the human genome, out of all the material recorded, and that's only 20nt long (not sure if they checked all genomes or just human. Maybe that chunk matches some small part of some other virus. You may occasionally find a single stretch of reads virus to virus that code for 6-12 amino acids, but this is very rare if they are not conserved, and if they are conserved, this becomes very obvious rather quickly across viruses).
I don’t see how, if I have 1000 chunks of 30 nt or more (Update: Turns out Contigs, which I think are what MPS techs produce, are 50-400nt long) could they EVER be rearranged into something else? You can't go 30nt without running into many deviations just across variants, let alone, 30 nt, 1000 times. And even if it was possible : nanopores can take the whole virus for us. that's 1000nt - 30000nt, covering the whole genome many times, and we can see it matches very well to the reference. And back to the amino acids. To rearrange them however you want i.e. to make the SARS 1 spike into SARS 2, you'd have to be able to do it in only 3nt chunks, so you could pick what amino acids come in what order.
I’m rather sure people that say these things and make these misinterpretations, have never:
1. Looked closely at many viral genomes
2. Looked closely at many viral amino acid orders in proteins of viruses
3. Looked closely at the way in which primers, PCRs, polymerases, or sequencing technologies actually work
4. Looked closely at basic chemistry, biochemistry and physics, and thus don’t comprehend fundamental concepts like conservation of matter
Looked closely at nanopore technologies
Looked closely at anything that gave them cognitive dissonance, when they realized they couldn’t instantly comprehend hundreds of years of scientific literature in a single afternoon
Nothing written here should be taken as support for PCRs as diagnostic tools. The presence of RNA exactly where it is supposed to be stopped by the innate immune system, using primers that then amplify regions you don’t verify later, is not a viable diagnostic method by any means. It CAN be, but only if the individual using the tools, and the designer behind said tools, are honest and competent. It would take appropriate primer distribution that couldn’t amplify incorrect targets and a lack of human error, just to ensure viral identification accuracy, if sequencing isn’t done after (hence why PCRs are unsuitable black boxes).
What PCR does well, is amplify stuff between primers, i.e. molecules that are bound to other molecules I know or suspect to be there. In language form, it says: “I know x is there, or I think x is there, Can I bind to x? Yes. Then: amplify everything after x, y times (controlled by experimenter). Then Sanger says “I can bind to x. What is after x on each molecule I find in my sample? Order these by weight and thus exact molecular length, and tell me what is on the end of each piece”.
So, PCR by itself, = crap. PCR with good Sanger after, = proof of the presence of a reference genome, or something so close we can literally monitor every point a strand deviated from the reference (which is nowhere near as often as is made out to be):
This study reports an identity of 99.97% between the full genome sequence generated and the reference strain at the nucleotide level and a 99.95% at amino acid level. Other sequences sampled from GenBank as shown in Table 2 had identity of 99–100% at both nucleotide and amino acid levels. The sequence analysed is not 100% identical to the reference genome, but 99.97% identity shows that there is a minimal or insignificant variation as the virus spreads across several environmental regions, from China to Nigeria at the moment. However, this seemingly insignificant variation, needs to be further studied as its corresponding changes in the protein building block could result in a mutation that will alter the behaviour of the virus. Lu and colleagues [16] established that coronaviruses have an average evolutionary rate of about 10−4 base substitution site per year with mutations arising at every replication cycle however that has not been the case with SARS-CoV-2 which has remained majorly conserved even after 6 months of its outbreak [7].